|
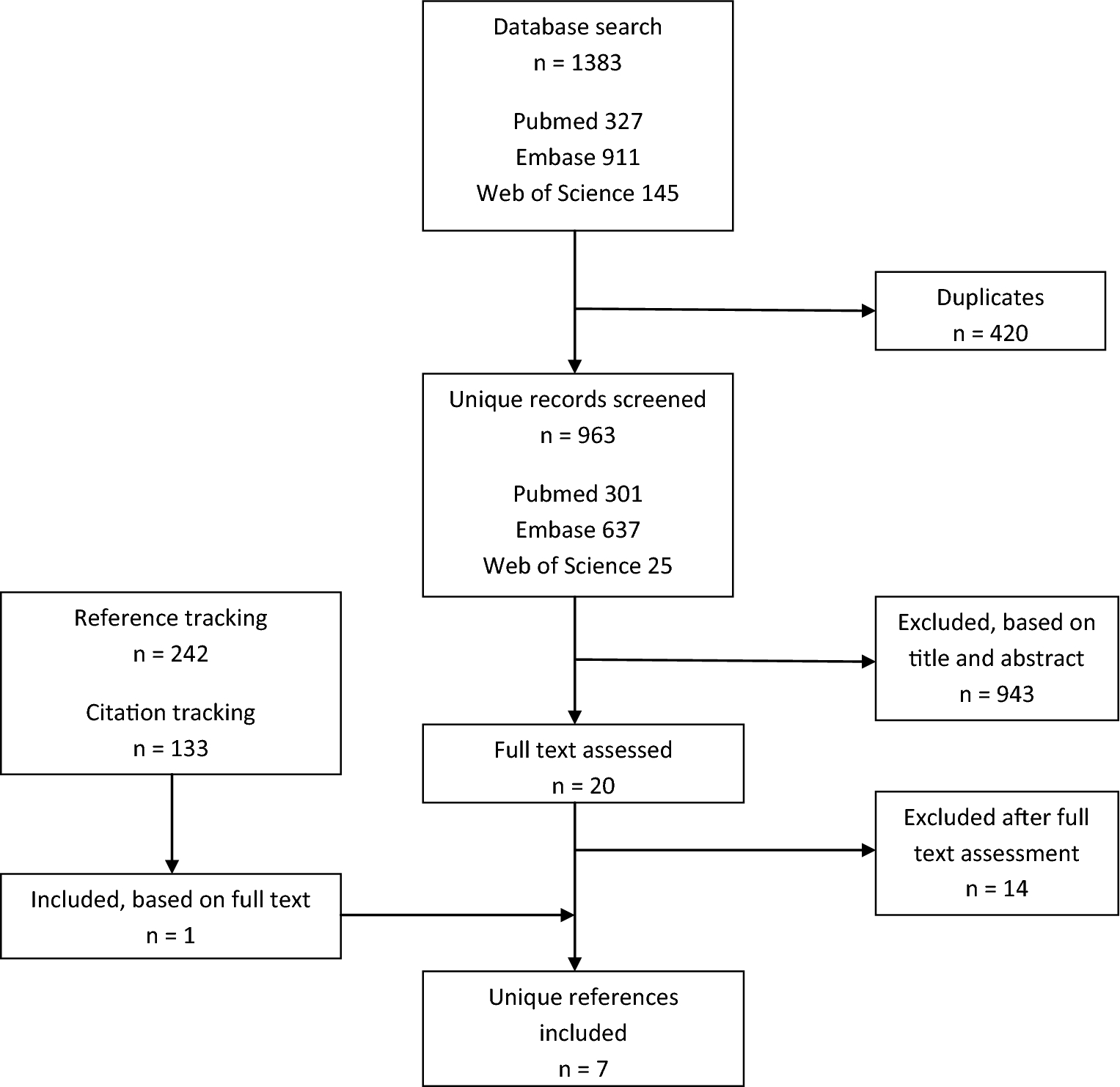
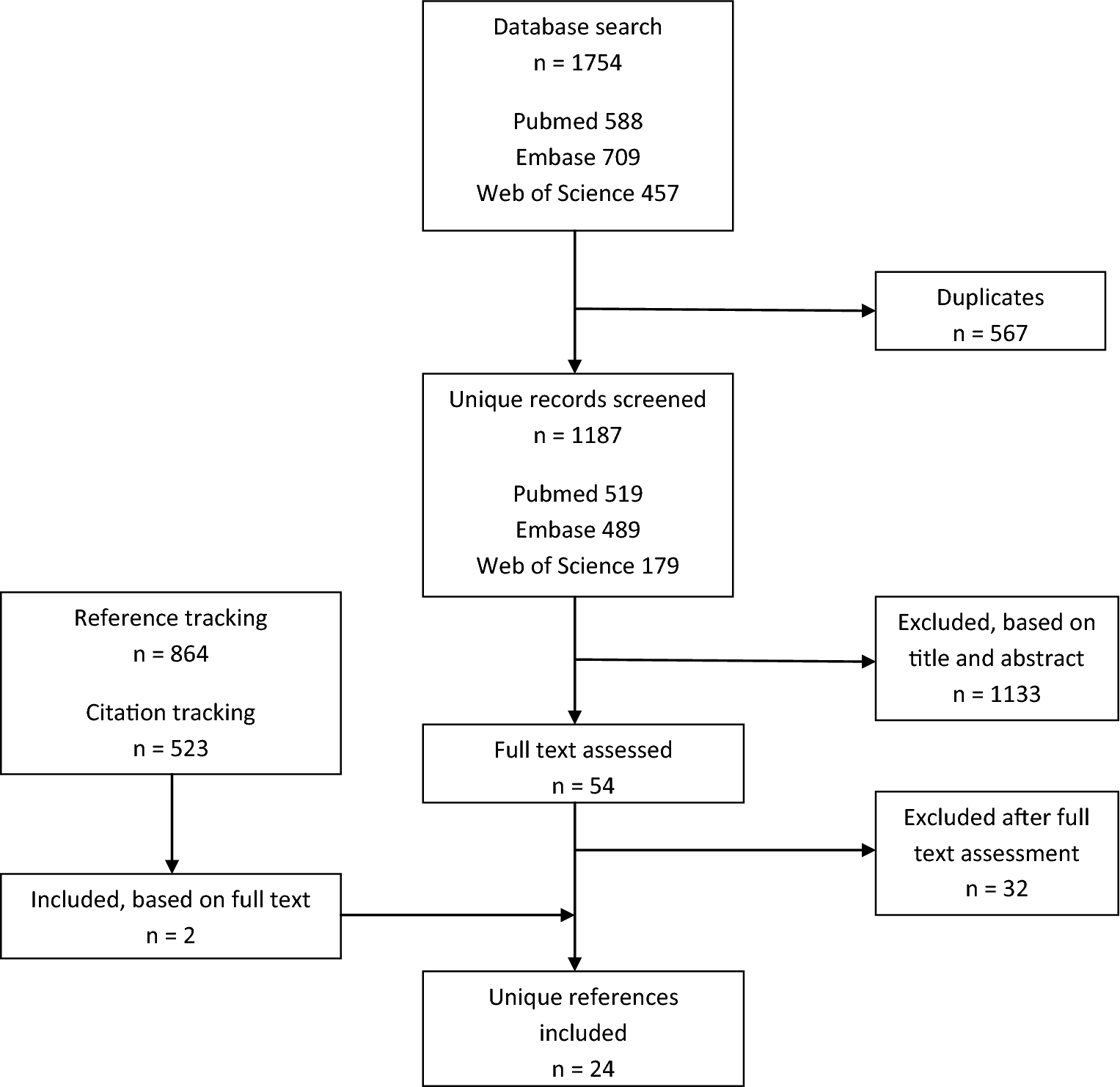
Bohman
|
Radical resection 29%, adjuvant external beam RT 42%
|
NR
|
NR
|
Older age, male sex, non-white race, earlier decade of diagnosis, tumour not confined to periosteum, tumour size ≥ 4 cm, no postoperative RT, no radical surgical resection, non-chondroid subtype
|
|
Uhl
|
Surgery 90% (biopsy 10%, R2 resection 90%—35% had recurrent disease). Adjuvant carbon ion RT 100%
|
Median 72 months (12–165)
|
Age ≥ 48, PTV > 75 ml, male gender, dose < 60 Gy, RT for recurrence
|
Older age, PTV2 > 75 ml, boost volume < 75 ml, RT after recurrent treatment
|
|
Weber
|
Surgery with curative intent prior to RT 100% (76% for initial disease; 24% for recurrent disease). Pencil beam scanning proton therapy 100%
|
Median 50 months (4–176) for both chordoma and chondrosarcoma
|
Optic apparatus compression, brainstem compression, GTV > 25 cc, recurrent disease, female gender, age > 40
|
Optic apparatus compression, brainstem compression, GTV > 25 cc, recurrent disease, female gender, age > 40
|
|
Terahara
|
RT 100% (98% combined proton/photon; 2% proton)
|
Mean 52 months, median 41 (5–174)
|
Male gender, older age, histology, lower minimum dose, bigger target volume, lower EUD, lower D5cc, other dosimetric parameters
|
NR
|
|
Wu
|
Prior surgery 21.5%, prior RT 13.9%, surgery 100%, adjuvant RT 50.6% (< 3 cm residual tumour GKRS, others LINAC)
|
Mean 63.9 months (10–158)
|
Prior operation history, prior RT history, non-total resection, hard tumour consistence, tight adherence to vital structure, extensive tumour location, stage III tumour, dedifferentiated subtype
|
Prior operation history, prior RT history, non-total resection, hard tumour consistence, tight adherence to vital structure, extensive tumour location, stage III tumour, dedifferentiated subtype
|
|
Ouyang
|
Surgery 100% (for primary resection group: total or near-total resection 33%, subtotal resection 48%, partial resection 12%), adjuvant RT 33% (< 3 cm residual tumour GKRS, others LINAC)
|
50.4 months (6–142)
|
Smaller extent of resection, dedifferentiated subtype
|
Smaller extent of resection, dedifferentiated subtype
|
|
O’Connell
|
Surgery 100%, adjuvant combined proton/photon RT 100%
|
Median 69 months (20–158)
|
Female gender, age > 40, volume > 70 ml, non-chondroid subtype
|
Female gender, age > 40, volume > 70 ml, non-chondroid subtype
|
|
Forsyth
|
Subtotal resection 78%, biopsy only 22%, adjuvant RT 76%
|
Median 99.6 months (67.2–356.4)
|
Age ≥ 40, bad neurological function, symptom duration prediagnosis > 365 days, male gender, headache, diplopia, papilledema, extraocular motor palsy, biopsy or shunt only, no RT
|
Age ≥ 40, bad neurological function, symptom duration prediagnosis > 365 days, male gender, headache, diplopia, papilledema, extraocular motor palsy, biopsy or shunt only, no RT
|
|
Boari
|
Prior surgery 22%, prior biopsy 18%, prior RT 7%, prior chemotherapy 7%. GTR 42%, subtotal resection 40%, partial resection 13%, biopsy 5%, adjuvant proton RT 42%, adjuvant fractionated RT 16%, adjuvant Gamma Knife RT 18%, adjuvant imatinib mesylate 4%
|
Mean 76 months (1–240)
|
Age > 48, rhinopharynx invasion, female gender, previous surgery, brainstem compression, cavernous sinus invasion, dural involvement, classic tumour histology, less than subtotal tumour resection, no adjuvant RT, preoperative KPS < 80
|
Age > 48, rhinopharynx invasion, female gender, previous surgery, brainstem compression, cavernous sinus invasion, dural involvement, classic tumour histology, less than subtotal tumour resection, no adjuvant RT, preoperative KPS < 80
|
|
Noël
|
Complete resection 17%, incomplete resection 71%, only biopsy 12%, adjuvant fractionated proton RT 100%
|
Median 34 months (3–74)
|
Histological subtype, older age, lower minimum dose delivered, lower dose delivered to 95% of the GTV, other dosimetric parameters
|
Failed local control, older age, dosimetric parameters
|
|
Yasuda
|
Previous surgery 42.5%, previous RT 45%, preoperative occlusion of involved vessel 12.5%, surgery 100% (43% radical, 48% subtotal, 10% partial), adjuvant RT 75% (43% proton/photon, 25% proton, 5% photon)
|
Mean 46.1 months, median 56.5 (3–99)
|
Age ≥ 47, male gender, CCJ tumour, recurrence surgery group, non-extensive resection, non-chondroid subtype
|
Age ≥ 40, male gender, CCJ tumour, recurrence surgery group, non-extensive resection, non-chondroid subtype
|
|
Cheng
|
Total resection 30%, subtotal resection 22%, partial resection 48%, adjuvant external beam RT 57%
|
Mean 85.2 months (18–288)
|
Lower tumour localisation, older age, bigger tumour, male gender, rectal invasion, longer duration of symptoms, tumour extension beyond cortical bone
|
Lower tumour localisation, older age, bigger tumour, male gender, rectal invasion, longer duration of symptoms, tumour extension beyond cortical bone
|
|
Schwab
|
Previous surgery 29%, surgery 100% (wide resection 63%, marginal resection 7%, contaminated margin 19%, intralesional resection 9%), adjuvant cryosurgery 14.3%, adjuvant external beam RT 36%
|
Mean 46 months (1–169)
|
Prior resection
|
Local recurrence, metastasis, prior resection, high-grade lesion, male gender, age > 65, tumour > 8 cm
|
|
Chen KW
|
Preoperative tumour embolisation 100%, surgery 100%, adjuvant RT 41.7%
|
Mean 74.4 months (16–182)
|
Age > 50, male gender, tumour size > 9.15 cm, tumour location above S3, surrounding muscle invasion, non-wide surgery, non-wide margins, no adjuvant RT
|
NR
|
|
Dubory
|
Previous intralesional surgery 14%, preoperative RT 7%, preoperative imatinib mesylate 14%, surgery 100% (wide resection 62%, marginal resection 21%, intralesional resection 14%), adjuvant RT 55%, adjuvant chemotherapy 3%
|
77.9 months (0–241)
|
Postoperative local infection, postoperative RT
|
Tumour size > mean, positive surgical margins
|
|
Mima
|
RT 100% (70% carbon ion, 30% proton)
|
Median 38 months (7–78)
|
Female gender, dose fractionation, ion type, tumour volume ≥ 400 ml
|
Tumour volume ≥ 400 ml, female gender, dose fractionation, ion type
|
|
Mukherjee
|
Surgery 87.7%, (adjuvant) RT 46.5%
|
NR
|
NR
|
Distant metastasis
|
|
Stacchiotti
|
Previous surgery 22.4%, surgery 100% (wide resection 35%, marginal resection 25%, intralesional resection 34%, inoperable 6%), adjuvant RT 31%, adjuvant anthracyclines/platinum-based chemotherapy 8%, adjuvant imatinib mesylate 11%
|
Median 142 months (IQrange 76–210)
|
Older age, bigger tumour, intralesional margins, sacral tumour, no RT, previously treated elsewhere
|
Older age, bigger tumour, intralesional margins, sacral tumour, no RT, previously treated elsewhere
|
|
McGirt
|
Surgery 100%, adjuvant RT 37%
|
≥ 60 months
|
NR
|
Older age, earlier year of diagnosis, sacral tumour, radiation therapy, increasing extent of invasion, more recent year of surgery, presence of metastasis, male gender, non-white race
|
|
Bergh
|
Previous surgery 26%, previous RT 5%, surgery 100% (wide resection 59%, marginal resection 15%, intralesional resection 26%), adjuvant RT 49%
|
Mean 97.2 months (1.2–276)
|
Sacral tumour, morphologic diagnostic procedure, older age, male gender, invasive diagnostic procedure outside current centre, previous surgery elsewhere, inadequate margins at initial surgery, inadequate margins at definitive surgery
|
Sacral tumour, morphologic diagnostic procedure, older age, male gender, invasive diagnostic procedure outside current centre, previous surgery elsewhere, inadequate margins at initial surgery, inadequate margins at definitive surgery, larger tumour size
|
|
Chen YL
|
Definitive combined high-dose proton/photon therapy (100%)
|
Mean 74.2 months, median 56 months (17.5–171.7)
|
Male gender, sacral tumour
|
Tumour volume > 500 cm3
|
|
Jawad
|
Surgery only (48%), RT only (10%), surgery and RT (33%), no therapy (9%)
|
NR
|
NR
|
Age > 59, male gender, non-white race, non-hispanic ethnicity, distant stage of disease, size > 8 cm, sacral tumour, no surgery, no RT, earlier year of diagnosis
|
|
Thieblemont
|
Surgery only (53.8%), surgery and RT (46.2%), adjuvant chemotherapy (26.9%)
|
NR
|
Female gender, age ≥ 60, sacral tumour
|
Male gender, age ≥ 60, sacral tumour
|
|
Meng
|
Previous surgery 22%, preoperative embolisation 42%, surgery 100% (en bloc resection 38%, total piecemeal resection 52%, subtotal resection 10%), local treatment with cisplatin or methotrexate after surgery 70%, adjuvant RT 9%
|
Mean 72 months, median 57 months (1–279)
|
Tumour in C1–C2 or S1–S5, Frankel score A–C, Enneking stage II–III, dedifferentiated subtype, Tomita score IV–VI, subtotal resection, increased intraoperative blood loss, no local treatment with cisplatin or methotrexate after surgery, no adjuvant RT, male sex, KPS < 80, prior treatment, distant metastasis, bladder and bowel dysfunction, tumour > 6 cm
|
Older age, male sex, KPS < 80, prior treatment, distant metastasis, bowel and bladder dysfunction, Frankel score A–C, Enneking stage II–III, Tomita score IV–VI, tumour > 6 cm, tumour in C1–C2 or S1–S5, subtotal resection, dedifferentiated subtype, no preoperative embolisation, increased intraoperative blood loss, no local treatment with cisplatin or methotrexate after surgery, no adjuvant RT, complication after surgery, postoperative recurrence, postoperative distant metastasis
|