Adult Paget's Disease of Bone: A Review
Abstract and Introduction
Abstract
Adult PD of bone is the second commonest metabolic bone condition after osteoporosis. The condition is characterized by increased bone cell activity, with bone-resorbing osteoclasts often larger and containing more nuclei than normal, and osteoblasts producing increased amounts of disorganized bone. This leads to expanded bone of poor quality possessing both sclerotic and lytic areas. PD of bone has a strong genetic element, with a family history being noted in 10–20% of cases. A number of genetic defects have been found to be associated with the condition. The most common disease-associated variants identified affect the SQSTM1 gene, providing insights into disease aetiology, with the clinical value of knowledge of SQSTM1mutation status currently under active investigation. The diagnosis may be suggested by an isolated raised total ALP without other identifiable causes. This can be confirmed on plain X-rays and the extent determined by isotope bone scan. The mainstays of treatment are the bisphosphonates, especially i.v. zoledronate, which results in long-term suppression of bone turnover. ALP is the usual means of monitoring the condition, although more specific bone turnover markers can be helpful, especially in coincident liver disease. Patients should be followed up to monitor for biochemical relapse or development of complications, which may require medical or surgical intervention.
Introduction
Adult PD of bone (PDB) was first described by Sir James Paget in 1877, a condition he referred to as osteitis deformans.[1] It is characterized by increased bone cell activity that results in expanded bone possessing both sclerotic and lytic areas. It is initiated by enhanced resorption by abnormal multinucleated osteoclasts followed by disorganized bone formation by osteoblasts.[2] There is a high rate of bone remodelling at affected sites in the skeleton, and the bone is highly metabolically active and has a high blood flow, which can make the overlying skin feel warm to the touch.[3] PDB is the second most common metabolic bone disorder after osteoporosis. Despite this, it is both underdiagnosed and undertreated, with many patients never coming to medical attention.[4] The condition tends to affect an older population, being unusual in individuals under 40 years of age. It can affect anything from a single bone (monostotic) to many bones (polyostotic).[2,3]The condition is often asymmetric with, for example, only one femur involved. PDB does not generally spread from bone to bone, and it is unusual for new bones to become involved.[5]
Epidemiology
PDB is more common in males than females; for example, in a study published in 2002, the incidence was reported as 5.4 cases/10 000 person-years in women of ⩾85 years, compared with 7.6 cases/10 000 person-years in men of the same age.[4] UK and other populations of British descent have the highest prevalence of PDB, with up to 2% in those over 55 years estimated to be affected (based on analyses of radiographic records over the period 1993–95). In contrast, PDB is rare in Scandinavia, the Indian subcontinent and the Far East.[6] Curiously in some countries, including the UK, the incidence and severity of PDB has declined rapidly in recent years;[7]in certain populations, incidence rates may only be 10–20% of those seen 20–30 years ago.
Pathogenesis
In PDB there is loss of the normal regulation of bone resorption and formation, with the process biased towards one or the other depending on the phase of the disorder.[8] There are three phases. The first is a lytic phase in which normal bone is resorbed by numerous, enlarged and more nucleated than normal osteoclasts and bone turnover is markedly increased. Secondly, a mixed phase of lytic and blastic activity characterised by rapid increases in bone formation from numerous osteoblasts. The newly synhesised bone is abnormal with collagen fibres deposited in a haphazard way. There is both osteobalstic and osteoclastic activity, but formation becomes dominant. Finally, there is sclerotic phase in which bone formation predominates, but the formed bone is disorganised (woven bone) and is weaker. The resultant bone has altered and often abnormal architecture. While PDB has previously been regarded as primarily a disease of osteoclasts, there is evidence that the functioning of stromal cells and osteoblasts is also abnormal, and there is loss of the usual tight regulation of the process of bone resorption and formation, for example, mediated via TGF-β.[9,10]
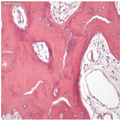
In the early phase of PDB, microscopic assessment can show a combination of bone resorption, endosteal fibrosis and prominent vascular sinusoids (Figure 1). There are increased numbers of enlarged osteoclasts with more nuclei than normal and increased bone resorption. Subsequently, there is increased formation of new bone when the characteristic microscopic features of PDB occur. The irregular and variably sized resorption bays (Howship lacunae) left by the excess osteoclast activity become filled in with bone newly synthesized by osteoblasts. As bone remodelling is not properly regulated, new bone is present in variably sized patches separated by prominent, irregular, scalloped cement lines, and referred to as a mosaic. Characteristically, a high proportion of the initial new bone formed is actually of lamellar type, but it is irregularly and randomly organized, therefore decreasing strength and load-bearing capacity.[11] In the later stages of PDB, the histology is that of thickly reconstructed bone with increased numbers of prominent and irregular cement lines that are prone to fracture. In the final sclerotic phase in which bone formation predominates, the collagen fibres are deposited in a haphazard way, the bone is disorganized (appearing woven) and is weaker. There is a modest increase in the incidence of osteosarcoma,[12,13] which should be distinguished from the periosteal (juxtacortical) bone masses that can be seen in PDB.
Figure 1.
Typical histology of adult PD of bone
An H and E image viewed down a microscope of a thin section of involved bone. It shows in more detail that, as the phases of resorption and formation progress, the trabeculae become abnormally shaped. There are numerous reversal lines, which make the trabeculae appear to be composed of several pieces fitted together—the so-called mosaic pattern.
Genetic factors play an important role in PDB, with 10–20% of patients having a family history of the disorder. Susceptibility probably arises from a combination of relatively rare pathogenic mutations and more common small-effect variants (that predispose to disease) at several loci within or close to genes relevant to osteoclast function.[14] The most well-characterized genetic links are mutations affecting the SQSTM1 gene, which encodes the SQSTM1/p62 protein (Sequestosome-1) and is found in up to 50% of familial and some sporadic cases of PDB.[15,16] Patients with SQSTM1mutations often have severe PDB, are diagnosed earlier and have a high degree of penetrance with increasing age.[17] The SQSTM1/p62 protein is an important regulator of osteoclast RANK-mediated NF−κB signalling, with mutation-associated activation of this pathway likely linked to disease aetiology.[18] Indeed, SQSTM1 mutation status plays a major role in determining the disease phenotype in patients, with other risk alleles associating in an additive manner.[19] Interestingly, SQSTM1 mutations are also linked to neurodegenerative disorders such as the amyotrophic lateral sclerosis and frontotemporal lobar degeneration spectrum, which exceptionally rarely can also coexist with PDB,[20] and PDB can also occur as part of the multisystem disorder inclusion body myopathy with PDB and frontotemporal dementia,[21] caused by mutations affecting the VCP gene. It is important to note that any rare neurodegenerative phenotypes are distinct from neurological symptoms that may result from neurological complications in PDB cases in which there is involvement of the spine or cranium. Severe PDB associated with giant cell tumour has also recently been linked to mutations affecting the gene encoding the zinc finger protein 687 (ZNF687), which like SQSTM1/p62 plays a role in NF−κB signalling.[22] Finally, variants in the OPTN gene are associated with PDB and in mouse models these result in increases in RANK ligand (RANKL)-dependent NF-κB activation.[23]
The declining incidence,[7] coupled with the fact that not everyone with gene defects develops PD, suggests that environmental factors must also play a role influencing development and severity of the disease. Viruses such as the measles virus have been proposed as an aetiologic factor, although involvement has not been confirmed in all studies.[24] There are probably other unknown environmental triggers.[25]
Clinical Features, Complications and Presentation
The clinical presentation and complications of PDB are a result of the increased bone cell activity in affected bone, bone expansion, deformity and poor bone quality (Table 1). In order of frequency, PDB affects the femur, spine, skull, sternum and pelvis, but can be found in any bone in the body.[26] Many patients are asymptomatic, and the diagnosis is often incidental as a result of blood tests revealing an isolated raised ALP, or imaging undertaken for other reasons. The serum calcium and phosphate are usually normal, although it is possible, albeit very rarely, for hypercalcaemia to occur during immobilization or should concomitant hyperparathyroidism develop. Subsequent X-rays and an isotope bone scan may then confirm the diagnosis and extent of the disease.
Bone pain occurs in ~50% of cases presenting clinically.[27] The pain may arise from the affected bone itself, when it is often described as lancinating[27,28] (occurring throughout the day, worse at night and on standing) in weight-bearing limbs.[26] Alternatively, it can arise from the altered biomechanics of limb deformity; for example, bowed tibia or femur, which alters gait and puts stress on joints and soft tissues and causes secondary OA.[2,29] Approximately 10–30% of PDB patients sustain fractures.[2] These may initially be incomplete, traversing the cortex (especially the outer borders) of bowed bones (fissure fractures). Fissure fractures predominantly, but not exclusively, affect weight-bearing bones, which are at high risk of complete fracture. Complete fractures are often transverse when they are referred to as chalk stick or banana. Bone healing seems to take place normally.[2] The development of osteosarcomas within the affected bone, or more rarely chondro- or fibrosarcomas, can also cause pain, a soft tissue mass and a rise in ALP level.[2,4]
Neurological complications occur generally as a result of bony overgrowth leading to compression, and the commonest is hearing loss, probably due to cochlear damage,[30] most famously in the composer Ludwig van Beethoven.[31] Compression at the base of the skull may lead to basilar invagination, cerebellar dysfunction and even obstructive hydrocephalus, resulting in nausea, ataxia, incontinence, altered gait and dementia. Thoracic and lumbar spine involvement may cause nerve entrapment and even spinal canal stenosis, but sometimes this is the result of the increased vascularity causing vascular steal phenomena. If this presents with paraparesis or paraplegia then this should be managed as an emergency (see below).
The increased vascularity can result in excessive blood loss should the bone fracture or any surgery be undertaken upon it unless the PDB has been brought under control beforehand. It could also potentially cause increased cardiac output and high-output cardiac failure should the disease be extensive and uncontrolled. Hultgren reported that there was an association between PDB and aortic stenosis, arteriosclerosis and intracardiac calcification.[32]
Diagnosis
The possibility of PDB is often first raised by an isolated raised ALP, which isoenzymes reveal to be largely of bone origin. Other possible causes of this need to be excluded, including vitamin D deficiency, hyperparathyroidism, hyperthyroidism, renal osteodystrophy and malignancy. Bone metastases, including prostate cancer and myeloma, can usually be differentiated by blood tests, plain X-rays and other imaging. Very occasionally it is necessary to resort to bone biopsy to be certain, especially if PDB and malignancy are found to be coexistent.
Bone turnover markers (BTMs) are not specific, and the diagnosis is best established by imaging. Isotope bone scans are very sensitive for detecting areas of increased bone cell activity—allowing the extent of disease to be determined—but are not specific. X-rays of affected bone will usually be diagnostic and show the characteristic features of the illness (Figure 2 and Figure 3). The three phases of PDB are evident on X-rays:[2,33] an early lytic phase giving the characteristic blade-of-grass appearance, progressing through the bone at about 8 mm/year;[34] a mixed picture of osteolysis and osteosclerosis; and finally a phase in which sclerosis is the main appearance.
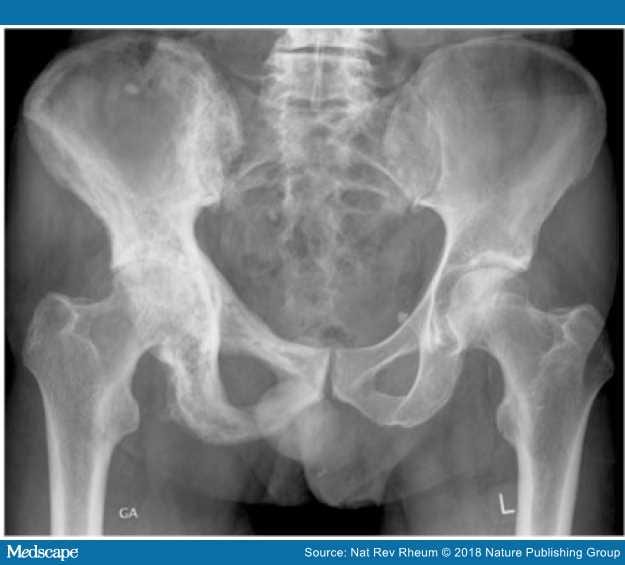
Figure 2.
Gentleman found on routine blood tests to have an isolated raised ALP
X-rays confirmed the presence of PD of bone affecting the right hemipelvis, where there is sclerosis. A subsequent isotope bone scan confirmed the disease to be confined to the pelvis.
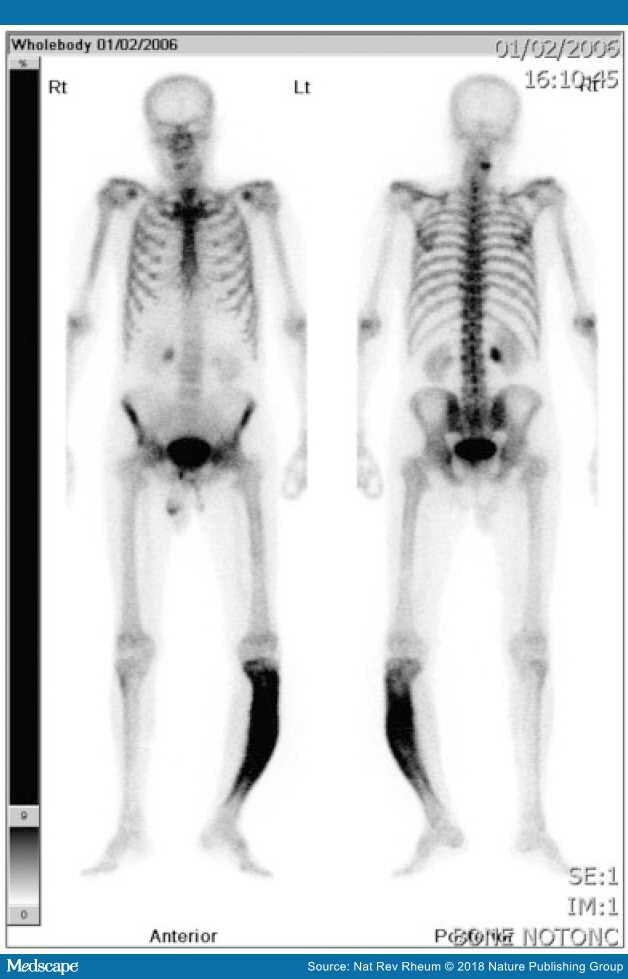
Figure 3.
Isotope bone scan of man with PD of bone affecting his left tibia
It is worth noting that the bowing has resulted in him standing on tiptoe. He had leg length discrepancy, with pain from the Pagetic bone, altered biomechanics and secondary OA.
The changes can also be detected with CT scans, MRI and PET CT, which can be useful in distinguishing from other potential causes and to assess complications.[3]
Treatment
The pharmacological treatment of PDB is targeted at reducing osteoclast function (Table 2). Other treatment is aimed at relieving symptoms and complications of the illness. Pain from PDB itself will improve with treatment, but often arises from complications, for example, secondary OA. Analgesia may start with paracetamol and escalate to more powerful agents; it may respond well to NSAIDs. Pain from nerve compression may be helped by medications such as amitriptyline, gabapentin or pregabalin. It is important to ensure that patients are vitamin D replete (⩾50 nmol/l), have good calcium intake and normal calcium levels prior to specific therapy, otherwise hypocalcaemia can be induced.
Bisphosphonates
The advent of potent bisphosphonates has revolutionized treatment. The current first-line treatment for active disease or in those at risk of complications is i.v. 5 mg zoledronate because of its potency and prolonged duration of action. Zoledronate brings about rapid normalization of BTMs and maintains this for many years.[2,26,35] It is also hoped, although the evidence is lacking, that treatment might prevent the development of complications.
Zoledronate has been studied in a randomized clinical trial comparing a single i.v infusion of 5 mg with risedronate.[36] After 6 months, 96% of patients receiving zoledronate had a therapeutic response compared with just 74% of those randomized to risedronate (P < 0.001), with ALP levels normalizing in 89 and 58% of patients, respectively (P < 0.001). At 2 years the response was 98% vs 57%,[37] and by 6 years this was 87 and 38%, respectively.[38] The zoledronate group showed a more rapid response and superior effects on quality of life, including pain. Zoledronate is well tolerated, although there is a 20–25% risk of flu-like reactions, which can be reduced by the use of paracetamol or NSAIDs. The acute-phase response can include uveitis in ~1% of patients, which requires prompt treatment and resolves quickly with topical steroids. Zoledronate is also potentially nephrotoxic and should not be given if the estimated glomerular filtration rate is <35 ml/min.
Both alendronate and risedronate have been studied in clinical trials. Alendronate normalized ALP in 60–70% of cases and was associated with healing of lytic lesions and restoration of normal lamellar bone histology.[39,40] Oral risedronate shows evidence of pain relief and normalization of ALP in 73% of patients.[41–44] Intravenous pamidronate has largely been replaced by the more effective bisphosphonates, but it is capable of normalizing ALP levels and sustaining the response for ⩾12 months.[45,46]
Alternatives
As a potent inhibitor of osteoclasts, Denosumab would seem to be an attractive alternative medication in those unable to have bisphosphonates. Unfortunately, experience is anecdotal at present, with only a handful of case reports and no evidence on the optimum dose, frequency and duration of affect..[47,48] Schwarze et al. used it in a case in which renal failure precluded the use of bisphosphonates.[47] Reid et al.[48] used Denosumab when an individual had severe musculoskeletal pain with alendronate. They found a good response, but ALP rose gradually after the first dose and symptoms reoccurred, necessitating a second dose 15 months after the first. They felt it to be less effective than i.v. bisphopshonates and suggested that the osteoclastic overactivity in PDB was not wholly mediated by RANKL.[48]We have found 60 mg s.c. Denosumab given every 6 months to work well at keeping the ALP suppressed when oral or i.v. bisphosphonates could not be used.
Management of Complications
Bisphosphonates are excellent at inducing biochemical remission. However, patients can be biochemically well controlled, yet still have pain or problems from complications, for example, secondary OA. Practical difficulties resulting from deformity and abnormal biomechanics can be helped by physiotherapy, orthotics and occupational therapy.
Orthopaedic Surgery
Orthopaedic surgery may be required for: correction of deformity, joint replacement, prophylactic nailing to prevent imminent fracture, and nerve compression. Pagetic bone activity should be brought under control with medical therapy prior to surgery in order to reduce vascularity and minimize the risk of excessive blood loss. Unless there are contraindications, 5 mg i.v. zoledronate should be given 1–2 months prior to surgery. If an oral bisphosphonate is to be used, then 3–4 months should be allowed for the medication to take effect before surgery.[27] Pre-treatment should also reduce the chances of post-operative complications, for example, loosening of the prosthesis or rapid progression of the Pagetic bone.[51]
Fissure fractures sometimes respond to treatment with bisphosphonates,[52]but there is a risk of complete fracture, and an orthopaedic opinion should be sought for consideration of prophylactic nailing. Osteotomy may be needed to correct deformity (e.g. for bowing of the tibia), or joint replacement may be needed for secondary OA in neighbouring joints. The indications for joint replacement are the same as in patients without PDB, but the deformity and presence of abnormal bone requires careful surgical planning. Joint replacement is often very successful in relieving pain and improving mobility.
Neurological Complications
In general, hearing loss does not seem to respond to treatment of the PDB.[52] One study of calcitonin suggested that progression of hearing loss might be slowed over 5–8 years,[53] and there is a reported case of reversal with pamidronate.[54] Intensive therapy has not been found to improve hearing.[55] Cochlear implants have been tried, but there is limited experience.[56]
Neurosurgical intervention is sometimes necessary for problems arising from compressive phenomena, for example, spinal stenosis. Spinal stenosis can present with paraparesis or paraplegia and should be managed as an emergency. The individual should be treated with intravenous zoledronate, which will control the disease activity and reduce the chances of complications from excess bleeding should surgery be necessary. Sometimes, the cause is vascular steal from active Pagetic bone in vertebrae, which may well be reversed by the intravenous bisphosphonate.[2,57–59] If medical management should fail, then surgical opinions should be sought urgently.
Congestive Cardiac Failure
High-output cardiac failure has been described in untreated PDB, but is rare. The authors have encountered patients with heart failure that improved on treatment of the PDB with intravenous zoledronate. There have also been case reports of cardiac failure responding to treatment of PDB.[60]
Mailgnant Transformation
Malignant transformation to an osteosarcoma has a poor prognosis. Fortunately osteosarcomas are rare, affecting <1% of patients with the disease.[2] It is essential that they be referred to a specialist centre with an interest in sarcomas. Giant cell tumours have also been described—although usually benign they should still be referred.
Monitoring
The measurement of BTMs provides an objective means of assessing disease activity, response to treatment and biochemical relapse. Serum ALP is often elevated and falls after treatment. ALP correlates well with the amount of skeletal involvement on X-rays and activity on isotope bone scans.[2,26] Total ALP is also both readily available and relatively inexpensive. Total ALP is therefore recommended as the most commonly used marker to monitor PDB. However, it is unreliable in the presence of liver or biliary tract disease and can be within the normal range at presentation, particularly in monostotic disease when more specific BTMs would be advantageous.[25] The total ALP can be monitored within the normal range[28] with values pre- and post-treatment noted. Post-treatment isotope bone scans would show reduced radionuclide uptake, but would mean additional radiation.[61]
Bone-specific markers include: the formation markers bone-specific alkaline phosphatase (BSAP) and procollagen type 1 amino terminal propeptide (P1NP), and resorption markers urinary C-terminal telopeptide, serum β C-terminal telopeptide (sβCTX) and urinary N-terminal telopeptide (uNTX). Osteocalcin has not been found to be useful in monitoring PDB.[62–64] A recent meta-analysis examined the utility of ALP, BSAP, P1NP, sβCTX and uNTX in PDB and incorporated 17 observational studies and one clinical trial, totalling 953 patients.[3] Prior to treatment, all had a moderate to strong correlation with quantitative bone scintigraphy (range 0.58–0.80), with no significant difference between them. There was good correlation between markers, and all had good sensitivity to detect disease. The uNTX was the only one found to fully discriminate from the reference range in individuals with low scintigraphic disease activity. All decreased significantly after treatment and had a moderate to strong correlation with the change in bone scintigraphy, except BSAP (P = 0.047). The authors concluded that, overall, P1NP was the best-performing BTM. The review is limited by the relatively small number of studies that were heterogeneous, varying in size of population, type of bisphosphonates used, length of follow-up and which BTMs were evaluated. There were also different methods used to calculate the scinitigraphic indices, and none compared monostotic with polyostotic disease.[3] The serum CTX has both alpha and beta forms, and it is worth noting that serum CTX can show paradoxical responses in PDB because of altered isomerization between the two forms.
The markers of bone turnover take between 10 days and 2 months to fall after treatment with bisphosphonates,[3,26] and sβCTx can continue to fall for up to 6 months.[65] Bone resorption markers tend to fall sooner than formation markers,[3,26] with a nadir of ~10 days (compared with 2–3 months for ALP).[36] This is useful if early assessment of response is needed.
ALP remains the mainstay of biochemical assessment of activity, but P1NP, BSAP, urinary C-terminal telopeptide, sβCTX or uNTX can all be useful. Of the latter bone-specific markers, P1NP has some advantages, and the Endocrine Society recommended its use, as well as BSAP, especially in situations where the total ALP is not helpful.[26]
Long-term Outcomes
Potent bisphosphonates result in long-term suppression of bone turnover, and this can last at least 6 years with zoledronate.[38] It has also been observed that those patients who had a P1NP of <40 μg/l or a total ALP of <80 IU/l 6 months after treatment with zoledronate were found to have a >90% chance of not relapsing over the follow-up period. Furthermore, patients who received zoledronate had better quality of life measurements throughout follow-up than those randomized to risedronate.[38] The Endocrine Society guidelines recommend that the aim should be to eliminate disease activity when it is likely to cause complications such as deformity, fracture or neurological sequelae.[26] In addition to BTMs, the Endocrine Society guidelines also recommend the use of filling in of lytic lesions on repeat X-rays and normalization of bone scintigraphy to demonstrate elimination of disease activity.[26]
Improvements in bone turnover do not necessarily equate to improvements in quality of life. Langston et al. examined quality of life in 1324 patients with PDB recruited to the PRISM study (randomized trial of intensive bisphosphonate treatment versus symptomatic management in PDB). They found that bone pain, previous bisphosphonate therapy and increasing age were negative predictors of physical summary score on the Short-form 36 (SF36), but that ALP did not predict the score. They concluded that it was important to not just address the ALP in sufferers, but also quality-of-life issues.[66]
The PRISM study was designed to test the hypothesis that intensive therapy to keep the ALP within the normal range might improve outcomes. Patients were randomized to either symptomatic treatment (only given for bone pain not responding to analgesics) or intensive therapy (with repeated treatments given irrespective of symptoms, with the aim of keeping the ALP within the normal range). The intensive therapy group received treatment to keep the ALP within the normal range, or if normal to treat if serial measurements revealed a rising trend. The patients were followed up for a median of 3 years (range 2–5 years). The intensive treatment group had significantly lower ALP after the first 4 months of the study and remained lower throughout follow-up (P < 0.001). However, there was no difference between the groups in SF36 scores, overall body pain, Pagetic bone pain, hearing thresholds, fractures or the requirement for orthopaedic surgery.[55] The authors concluded that striving to normalize ALP with intensive therapy had no advantage over symptomatic treatment. These results were disappointing, but there could be a number of reasons for this. The majority of patients recruited had established disease, with 70% having had previous bisphosphonate treatment and 50% having normal ALP levels at entry. Also, the difference in proportion of patients with ALP within the normal range was relatively low, with between 17.6 and 21.5% more patients with normal ALP in the intensive group compared with in the control group. The study was also powered to detect a difference in fracture rates, with all the other outcomes being secondary end points. So it is possible that better results could be obtained with patients who were newly diagnosed and who had untreated, active disease at baseline. As established patients, some of their morbidity could be due to complications of the illness such as secondary OA, which will not respond to bisphosphonates.
PRISM also predates the use of i.v. zoledronate and only had oral bisphosphonates, pamidronate and calcitonin as available treatments. A 3-year extension to PRISM was undertaken to test the hypothesis that zoledronate might be more effective (PRSIM-EZ) in 502 subjects, of whom 270 continued the intensive therapy with zoledronate and 232 with symptomatic treatment. The ALP levels decreased further in the intensive group, but there were still no differences between the groups in quality-of-life scores or bone pain.[67] In contrast, a 6.5-year follow-up of the zoledronate versus risedronate study by Reid et al. found relapse rates were substantially higher in the risedronate group (23 of 115, 20%) compared with the zoledronate group (1 of 152, 0.7%, P < 0.001). The overall SF36 scores improved in the zoledronate group compared with those receiving risedronate, and these improvements were maintained throughout the 6.5 years of follow-up, whereas scores declined in the risedronate group (P < 0.01). The pain scores of the SF36 also improved in the zoledronate group at 24 [7.5 (2.6), P = 0.005] and 36 months [5.6 (2.4), P = 0.02), whereas there were no significant changes in the risedronate group at any time point.[37]
Long-term Follow Up
The initial assessment of biochemical and clinical response to treatment should take place between 3 and 6 months after treatment. Further follow-up will be needed to assess biochemical relapse and for potential complications. The advent of potent bisphosphonates such as zoledronate has allowed less frequent follow-up for assessing activity via BTMs to annually or biannually.[26,37] If less potent bisphosphonates have been used, then measuring BTMs every 6–12 months may be advisable. The BTMs will tend to rise over time, and a decision on when to re-treat will need to be made. There is much debate as to whether to treat only symptomatic individuals or to re-treat once the bone markers have risen to above the normal range, double post-treatment values or returned to pre-treatment levels. More evidence would need to be available before a definitive answer can be given to this question. Worsening or new pain in Pagetic bone (especially if this is biochemically well-controlled) requires assessment as to the cause and appropriate imaging.
Concluding Remarks
Adult PDB is a very common metabolic bone disorder, most frequently affecting men and women over 40 years of age. Although often asymptomatic, it can cause pain and result in considerable morbidity. It is uncommonly diagnosed, and even when identified it is poorly understood and inadequately treated, despite the efficacy of bisphosphonates. It is hoped that the use of intravenous zoledronate will reduce the frequency of bisphosphonate requirement to suppress the disease activity and possibly reduce long-term complications. There is a need for further research to better understand both the pathogenesis of the illness and prevention of long-term complications.
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877;60:37–64.
- LeBoff MS. Metabolic bone disease. In: Kelly WN, Ruddy SR, Harris, ED, Sledge C, eds. Textbook of rheumatology. Vol. 2, 5th edn. Philadelphia: W.B. Saunders, 1997: 1574–80.
- Al Nofal AA, Altayar O, Ben Khadra K et al. Bone markers in Paget's disease of the bone: a systemic review and meta-analysis. Osteoporosis Int 2015;26:1875–91.
- Van Staa TP, Selby P, Leufkens HG et al. Incidence and natural history of Paget's disease of bone in England and Wales. J Bone Mine Res 2002;17:465–71.
- Ethel SS, Roodman GD. Paget's disease of bone. In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7th edn. Washington DC: American Society for Bone and Mineral Research, 2008: 335–42.
- Takata S, Hashimoto J, Nakatsuka K et al. Guidelines for diagnosis and management of Paget's disease of bone in Japan. J Bone Miner Metab 2006;24:359–67.
- Cundy T, Reid IR. Reprint: Paget's disease of bone. Clin Biochem 2012;45:970–5.
- Siris ES. Paget's disease of bones and joints. J Bone Miner Res 1998;13:1061–5.
- Ralston SH. Pathogenesis of Paget's disease of bone. Bone 2008;43:819–25.
- Tang Y, Wu X, Lei W et al. TGF-beta1_induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 2009;15:757–65.
- Klein MJ, Bona SF, Freemont T et al. Miscellaneous disorders of ossification, calcification, cellular coupling and osteolysis. In: Klein MJ et al., ed. The AFIP atlas of nontumour pathology fascicle 9: non-neoplastic disease of bone. Vol. 9, 1st edn. Rockville, USA: American Registry of Pathology, 2011: 828–42.
- Hadjipavlou A, Lander P, Srolovitz H, Enker I. Malignant transformation in Paget's disease. Cancer 1992;70:160–9.
- Price CHG, Goldie W. Paget's sarcoma of bone. A study of 80. cases from the Bristol and Leeds bone tumour registries. J Bone Joint Surg 1969;51:205–24.
- Albagha OM. Genetics of Paget's disease of bone. Bonekey Rep 2015;4:756. doi:10.1038/bonekey.2015.125.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1(SQSTM1/p62) in Paget's disease of bone. Am J Hum Genet 2002;70:1582–8.
- Hocking LJ, Lucas GJ, Darosjewska A et al. Domainspecific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum Mol Genet 2002;11:2735–9.
- Ralston SH, Langston AL, Reid IR. Pathogenesis and management of Paget's disease of bone. Lancet 2008;372:155–63.
- Goode A, Long JE, Shaw B et al. Paget disease of boneassociated UBA domain mutations of SQSTM1 exert distinct effects on protein structure and function. Biochim Biophys Acta 2014;1842:992–1000.
- Albagha OM, Visconti MR, Alonso N et al. Common susceptibility alleles and SQSTM1mutations predict disease extent and severity in a multinational study of patients with Paget's disease. J Bone Miner Res 2013;28:2338–46.
- Almeida MR, Letra L, Pires P et al. Characterisation of an FTLD-PDB family with the co-existence of SQSTM1 mutation and hexanucleotide (G4C2) repeat expansion in C90rf72gene. J Neurobiol Aging 2016;40:191.
- Nalbandian A, Dunkervoort S, Dec E et al. The multiple faces of valosin containing protein associated diseases: inlcusion body myopathy, frontotemporal dementia and amyotrophic lateral sclerosis. J Mol Neurosci 2011; 45:522–31.
- Divisato G, Formicola D, Esposito T et al. ZNF687 mutations in severe Paget disease of bone asscoiated with giant cell tumor. Am J Hum Genet 2016;9892:275–86.
- Obaid R, Wani S, Azfer A et al. Optineurin negatively regulates osteoclast differentiation by modulating NFkB and interferon signalling; implication for Paget's disease. Cell Rep 2015;13:1096–102.
- Ralston SH, Afzal MA, Helfrich MH et al. Multicenter blinded analysis of RT-PCR detection methods for paramyxoviruses in relation to Paget's disease of bone. J Bone Miner Res 2007;22:569–77.
- Layfield R. The molecular pathogenesis of Paget's disease of bone. Expert Rev Mol Med 2007;9:1–13.
- Singer F, Bone HG, Hosking DJ et al. Paget's Disease of Bone: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014;99:4408–22.
- Tan A, Ralston SH. Clinical presentation of Paget's disease: evaluation of a contemporary cohort and systematic review. Calcif Tissue Int 2014;95:385–92.
- Tuck SP, Fordham JN. The current management of Paget's disease. Prescriber 2001;12:97–102.
- Frank WA, Bress NM, Singer FR, Krane SM. Rheumatic manifestations of Paget's disease of bone. Am J Med 1974;56:592–603.
- Monsell EM. The mechanism of hearing loss in Paget's disease of bone. Laryngoscope 2004;114:598–606.
- Oiseth SJ. Beethoven's autopsy revisited: a pathologist sounds a final note. J Med Biogr 2015; Advance Access published 27 October 2015, doi: 10.1177/09677720 15575883
- Hultgren HN. Oseitis deformans (Paget's disease) and calcific diseases of the heart valves. Am J Cardiol 1998;81:1461–4.
- Smith SE, Murphy MD, Motamedi k et al. From the archives of the AFIP. Rdaiological spectrum of Paget's disease of bone and its complications with pathological correllation. Radiographics 2002;22:1191–216.
- Maldague B, Malghem J. Dynamic radiological patterns of Paget's disease of bone. Clin Orthop Relat Res 1987;217: 126–51.
- Devogelaer JP, Bergmann P, Body JJ et al. Management of patients with Paget's disease: a consensus document of the Belgium Bone Club. Osteoporosis Int 2008;19: 1109–17.
- Reid IR, Miller P, Lyles K et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget's disease. New Eng J Med 2005;353:898–908.
- Hosking D, Lyles K, Brown JP et al. Long term control of bone turnover in Paget's disease with zoledronic acid and risedronate. J Bone Miner Res 2007;22:142–8.
- Reid IR, Lyles K, Su G et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011;26: 2261–70.
- Reid IR, Nicholson GC, Weinstein RS et al. Biochemical and radiological improvement in Paget's disease of bone treated with alendronate: a randomized, placebo controlled trial. Am J Med 1996;101:341–8.
- Siris E, Weinstein RS, Altman RD et al. Comparative study of alendronate versus etidronate for the treatment of Paget's disease of bone. J Clin Endocrinol Metab 1996;81:961–7.
- Hosking DJ, Eusebio RA, Chines AA. Paget's disease of bone: reduction in disease activity with oral rideronate. Bone 1998;22:51–5.
- Singer FR, Clemens TL, Eusebio RA, Bekker PJ. Risedronate, a highly effective oral agent in the treatment of patients with severe Paget' s disease. J Clin Endocrinol Metab 1998;83:1906–10.
- Siris ES, Chines AA, Altman RD et al. Risedronate in the treatment of Paget's disease of bone: an open label, multicenter study. J Bone Miner Res 1998;13:1032–8.
- Miller PD, Brown JP, Siris ES et al. A randomized, double blind comparison of risedronate and etidronate in the treatment of Paget's disease of bone. Paget's risedronate/etidronate study group. Am J Med 1999;106: 513–20.
- Siris ES. Perspectives: a practical guide to the use of pamidronate in the treatment of Paget's disease. J Bone Miner Res 1994;9:303–4.
- Gallacher SJ, Boyce BF, Patel U et al. Clinical experience with pamidronate in the treatment of Paget's disease of bone. Ann Rheum Dis 1991;50:930–3.
- Schwarze P, Rasmussen AQ, Kvist TM et al. Paget's disease of the bone after treatment with denosumab: a case report. Bone 2012;50:1023.
- Reid IR, Sharma S, Kalluru R, Eagleton C. Treatment of Paget's disease of bone with denosumab: case report and literature review. Calcif Tissue Int 2016;99:322–25.
- DeRose J, Singer FR, Avramides A et al. Response of Paget's disease to porcine and salmon calcitonins. Effects of long term treatment. Am J Med 1974;56:858–66.
- Doyle FH, Pennock J, Greenberg PB et al. Radiological evidence of a dose-related response to long-term treatment of Paget's disease with human calcitonin. Brit J Radiol 1974;47:1–8.
- Marr DS, Rosenthall DI, Cohen GL, Tomford WW. The rapid postoperative osteolysis in Paget disease. A case report. J Bone Joint Surg Am 1994;76:274–7.
- Siris ES, Roodman GD. Paget's disease of bone. In: Rosen CJ, Compston, JE, Lian JB, eds. Primer in the metabolic bone diseases and disorders of mineral metabolism. 7th edn. Washington DC: American Society for Bone and Mineral Research, 2008: 335–43.
- El Sammaa M, Linthicum FH, House HP, House JW. Calcitonin as treatment for hearing loss in Paget's disease. Am J Otol 1986;7:241–3.
- Murdin L, Yeoh LH. Hearing loss treated with pamidronate. J R Soc Med 2005;98:272–4.
- Langston AL, Campbell MK, Fraser WD et al. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget's disease of bone. J Bone Miner Res 2010;2591:20–31.
- Bacciu A, Pasanisi E, Vincenti V et al. Paget's disease and cochlear implantation. J Laryngol Otol 2004;118: 810–3.
- Herzberg L, Bayliss E. Spinal cord syndrome due to noncompressive Paget's disease of bone: a spinal artery steal phenomenon reversible with calcitonin. Lancet 1980;2:13–5.
- Chen JR, Rhee RS, Wallach S et al. Neurological disturbances in Paget disease of bone: response to calcitonin. Neurology 1979;29:448–57.
- Walpin LA, Singer FR. Paget's disease. Reversal of severe paraparesis using calcitonin. Spine 1979;4:213–9.
- Woodhouse NJ, Crosbie WA, Mohamedally SM. Cardiac output in Paget's disease: response to long-term salmon calcitonin therapy. Br Med J 1975;4:686.
- Avramidis A, Polyzos SA, Moralidis E et al. Scintigraphic, biochemical and clinical response to zoledronic acid treatment in patients with Paget's disease of bone. J Bone Miner Metab 2008;26:635–41.
- Coulton LA, Preston CJ, Couch M, Kanis JA. An evaluation of serum osteocalcin in Paget's disease of bone and its response to diphosphonate treatment. Arthritis Rheum 1988;31:1142–7.
- Randall AG, Kent GN, Garcia-Webb P et al. Comparison of biochemical markers of bone turnover in Paget disease treated with pamidronate and a proposed model for the relationships between measurements of the different forms of pyridinoline cross-links. J Bone Miner Res 1996;11:1176–84.
- Reid IR, Davidson JS, Wattie D et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget's disease of bone. Bone 2004;35:224.
- Alvarez L, Guanabens N, Peris P et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget's disease. Bone 2001;29:447–52.
- Langston AL, Campbell MK, Fraser WD et al. Clinical determinants of quality of life in Paget's disease of bone. Calcif Tissue Int 2007;80:1–9.
- Goodman K, MacLennan G, Selby P, Fraser W, Ralston S. Clinical outcome with long-term bisphosphonate therapy in Paget's disease of bone: the PRISM-EZ study. J Bone Miner Res 2013;28:884–431.